Appendice N — Diagnostica delle catene MCMC
Prima di procedere all’interpretazione dei parametri stimati da un modello in Stan, è essenziale verificare che il processo di campionamento MCMC si sia svolto in modo affidabile. Le catene generate possono infatti fornire risultati ingannevoli se non hanno esplorato adeguatamente lo spazio delle distribuzioni a posteriori.
Stan mette a disposizione un insieme articolato di indicatori diagnostici per valutare la qualità del campionamento. Questi permettono di rispondere a domande cruciali:
- Le diverse catene hanno convergito verso la stessa distribuzione stazionaria?
- La variabilità osservata nei campioni riflette fedelmente la vera distribuzione a posteriori?
- Esistono segnali che indicano regioni dello spazio dei parametri rimaste inesplorate?
Solo dopo aver accertato che il campionamento sia robusto e privo di anomalie significative, è possibile procedere in sicurezza all’analisi e all’interpretazione dei parametri stimati.
| Indicatore | Valore accettabile | Valore problematico |
|---|---|---|
| \(\hat{R}\) | < 1.01 | > 1.05 |
| ESS (bulk e tail) | > 400 | < 100 |
| Transizioni divergenti | 0 | > 0 |
| BFMI | > 0.3 | < 0.2 |
| Pareto \(\kappa\) (LOO) | < 0.5 | > 0.7 |
N.1 Un modello di riferimento
Per illustrare le procedure diagnostiche, utilizziamo un semplice modello beta-binomiale. Questo esempio permette di concentrarsi sugli strumenti diagnostici senza la complessità di modelli più articolati.
stancode <- "
data {
int<lower=1> N; // numero di prove
array[N] int<lower=0, upper=1> y; // esiti (0/1)
}
transformed data {
int<lower=0, upper=N> k = sum(y); // numero totale di successi
}
parameters {
real<lower=0, upper=1> theta; // probabilità di successo
}
model {
theta ~ beta(1, 1); // prior uniforme
k ~ binomial(N, theta); // verosimiglianza
}
generated quantities {
vector[N] log_lik;
for (n in 1:N) {
log_lik[n] = bernoulli_lpmf(y[n] | theta);
}
}
"mod <- cmdstanr::cmdstan_model(
cmdstanr::write_stan_file(stancode),
compile = TRUE
)fit <- mod$sample(
data = data_list,
iter_warmup = 1000,
iter_sampling = 4000,
chains = 4,
parallel_chains = 4,
seed = 4790,
refresh = 0
)N.2 Grafici di tracciamento
I grafici di tracciamento (trace plot) visualizzano la sequenza dei valori campionati per ciascun parametro lungo le iterazioni delle catene MCMC. Questa rappresentazione costituisce il controllo diagnostico preliminare più immediato e informativo.
N.2.1 Interpretazione di un trace plot ideale
Un grafico di tracciamento che indica un campionamento efficace presenta le seguenti caratteristiche:
- Miscelazione efficiente (good mixing): le catene oscillano in modo rapido e apparentemente casuale attorno a un livello stabile, assumendo l’aspetto visivo di un “rumore bianco”.
- Convergenza: le diverse catene (tipicamente rappresentate da colori differenti) si sovrappongono e si mescolano, indicando che tutte esplorano la medesima regione stazionaria dello spazio dei parametri.
- Stazionarietà: non sono visibili andamenti sistematici, derive nel tempo o cambiamenti nella dispersione lungo le iterazioni.
N.2.2 Segnali di potenziali problemi
Pattern che richiedono attenzione includono:
- catene che rimangono separate e non si sovrappongono;
- andamenti a gradini, salti improvvisi o persistenza in stati metastabili;
- strutture periodiche, cicli o altri pattern regolari che suggeriscono autocorrelazione eccessiva;
- deriva graduale verso valori superiori o inferiori.
mcmc_trace(fit$draws("theta")) +
labs(x = "Iterazione post-warmup", y = expression(theta))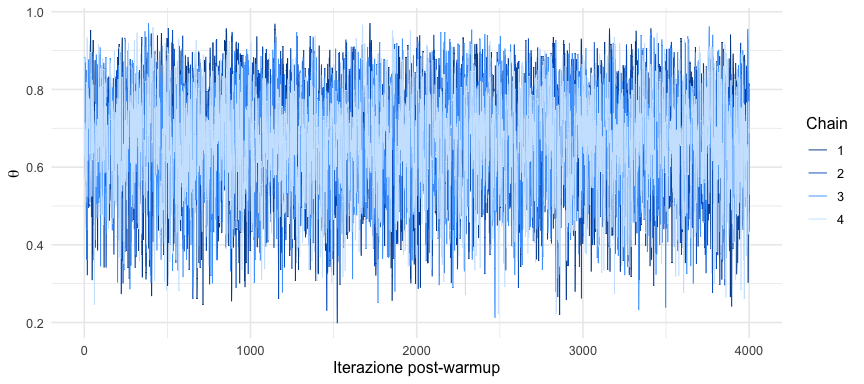
N.2.3 Strategie correttive per problemi di convergenza
Quando i trace plot rivelano anomalie, si possono considerare i seguenti interventi:
Aumentare il numero di iterazioni: un numero maggiore di campioni, eventualmente con un periodo di warmup più esteso, può permettere alle catene di superare periodi transitori e raggiungere la stazionarietà.
Rivedere le specifiche delle distribuzioni a priori: prior eccessivamente diffuse possono creare problemi di identificazione, mentre prior troppo informative possono vincolare le catene in regioni improbabili.
Riparametrizzare il modello: trasformazioni delle variabili (ad esempio, centratura, standardizzazione o passaggio a una scala logaritmica) possono semplificare la geometria dello spazio dei parametri e migliorare l’efficienza del campionatore.
Esaminare i dati di input: valori anomali, scale molto diverse tra le covariate o errori di codifica possono creare regioni di instabilità nella distribuzione a posteriori.
N.3 Grafici della densità posteriore
I grafici di densità mostrano la distribuzione dei valori campionati per ciascun parametro, offrendo una rappresentazione sintetica della distribuzione a posteriori marginale.
mcmc_dens(fit$draws("theta")) +
labs(x = expression(theta), y = "Densità")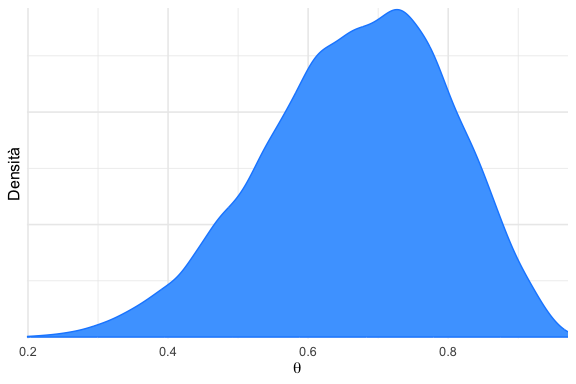
N.3.1 Caratteristiche di una densità posteriore ben stimata
Una densità che indica un campionamento adeguato è tipicamente:
- liscia e regolare, senza irregolarità o “rumore” nella curva;
- unimodale, presentando un singolo picco principale (salvo modelli che prevedano esplicitamente multimodalità);
- con code ben comportate, che decadono in modo graduale e senza interruzioni.
N.3.2 Segnali di potenziali problemi
Profili di densità che possono indicare problematiche includono:
- multimodalità inattesa, che può suggerire problemi di identificazione del modello o catene intrappolate in mode locali;
- code eccessivamente lunghe, piatte o irregolari, spesso indice di una scarsa informazione nei dati o di prior troppo deboli;
- aspetto frastagliato o a “denti di sega”, sintomo di un campionamento insufficiente o di un’eccessiva autocorrelazione che impedisce una rappresentazione fedele della distribuzione.
Un esame congiunto delle densità di tutte le catene consente inoltre di verificare che esse siano sostanzialmente sovrapposte, fornendo un ulteriore controllo visivo sulla convergenza.
N.4 Autocorrelazione
L’autocorrelazione quantifica la dipendenza statistica tra campioni successivi all’interno di una catena MCMC. Un’elevata autocorrelazione riduce l’informazione effettiva contenuta nei campioni, aumentando la varianza delle stime e diminuendo l’efficienza computazionale.
mcmc_acf(fit$draws("theta"), lags = 20)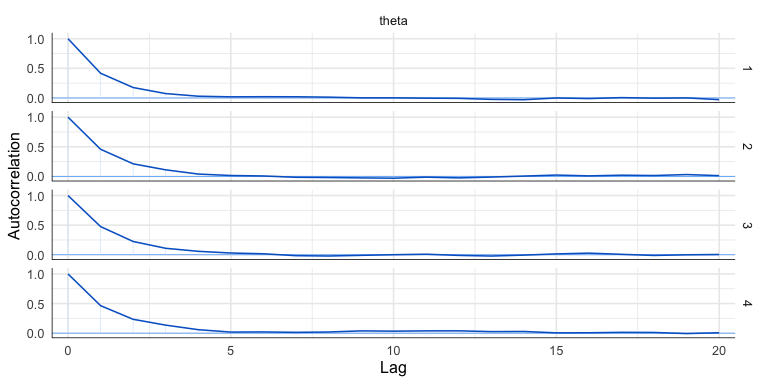
N.4.1 Interpretazione del grafico di autocorrelazione
- Autocorrelazione bassa: la funzione di autocorrelazione decade rapidamente verso zero, indicando che i campioni sono pressoché indipendenti. Questo è lo scenario ideale per un campionamento efficiente.
- Autocorrelazione elevata: valori significativi persistono anche a lag elevati, rivelando una forte dipendenza tra campioni distanti. Questo segnala un’inefficiente esplorazione dello spazio dei parametri e può invalidare le stime degli errori standard.
N.4.2 Strategie per ridurre l’autocorrelazione
Quando si riscontra un’elevata autocorrelazione, si possono adottare le seguenti contromisure:
Prolungare la durata del campionamento: un numero maggiore di iterazioni può compensare la ridotta informazione per campione, permettendo di ottenere un numero efficace di campioni indipendenti sufficiente.
Applicare il thinning: conservare solo un campione ogni \(k\) iterazioni (ad esempio, ogni 5 o 10) può ridurre la dipendenza nei dati conservati, sebbene questa pratica comporti uno spreco di informazioni e venga generalmente sconsigliata se non necessario per motivi di archiviazione.
Migliorare l’efficienza dell’algoritmo: aumentare il parametro
adapt_deltain Stan può produrre step-size più piccoli e un’esplorazione più conservativa. Spesso, la soluzione più efficace è una riparametrizzazione del modello per migliorare la geometria dello spazio dei parametri e facilitare il movimento del campionatore.
N.5 Dimensione effettiva del campione (ESS)
La dimensione effettiva del campione (Effective Sample Size, \(N_{\text{eff}}\)) misura l’informazione statistica indipendente contenuta in una catena MCMC autocorrelata. Espresso come equivalente numerico di campioni indipendenti e identicamente distribuiti (i.i.d.), quantifica l’efficienza del campionamento.
La formula di base è data da:
\[ N_{\text{eff}} = \frac{T}{1 + 2 \sum_{s=1}^{\infty} \rho_{s}}, \] dove \(T\) rappresenta il numero totale di campioni e \(\rho_s\) è l’autocorrelazione al lag \(s\). Valori elevati di autocorrelazione riducono sostanzialmente \(N_{\text{eff}}\).
N.5.1 ESS bulk ed ESS tail
In Stan si distinguono due diverse metriche ESS, ciascuna focalizzata su una regione specifica della distribuzione:
- ESS bulk: valuta l’efficienza del campionamento nella regione centrale della distribuzione (tipicamente attorno alla moda o alla media). È la misura rilevante per la stima di parametri di locazione come la media posteriore.
- ESS tail: misura l’efficienza del campionamento nelle code della distribuzione, fondamentale per la stima accurata di quantili estremi (ad esempio, il 2.5° e il 97.5° percentile per un intervallo credibile al 95%).
Per garantire una stima affidabile, sia l’ESS bulk che l’ESS tail per ogni parametro dovrebbero essere superiori a 400. Valori inferiori indicano che la catena potrebbe non aver esplorato adeguatamente la corrispondente regione della distribuzione.
# Visualizzazione del sommario con ESS
fit$summary(variables = "theta")# A tibble: 1 × 10
variable mean median sd mad q5 q95 rhat ess_bulk ess_tail
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 theta 0.668 0.678 0.132 0.137 0.436 0.868 1.00 5663. 6067.# Rapporto tra ESS e numero totale di campioni per tutti i parametri
ratios <- neff_ratio(fit)
mcmc_neff(ratios, size = 2) +
labs(title = "Rapporto ESS / N totale")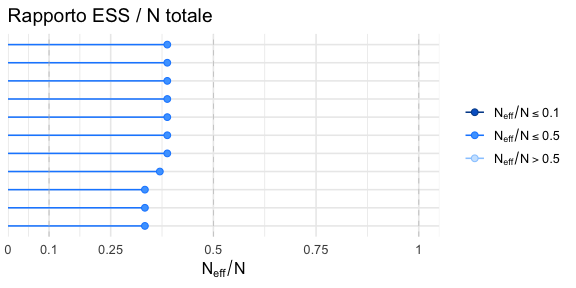
N.5.2 Interpretazione e intervento
Un rapporto \(N_{\text{eff}}/T\) basso (ad esempio, < 0.1) segnala un’elevata autocorrelazione e un’efficienza di campionamento scarsa. In questi casi, le strategie correttive sono le stesse indicate per il problema dell’autocorrelazione: aumentare il numero totale di iterazioni, migliorare la parametrizzazione del modello o ottimizzare i parametri di controllo dell’algoritmo HMC/NUTS.
N.6 La statistica \(\widehat{R}\) (R-hat)
La statistica \(\widehat{R}\), sviluppata da Gelman e Rubin, è una metrica fondamentale per diagnosticare la convergenza delle catene MCMC. Essa confronta la variabilità tra le diverse catene (\(B\)) con la variabilità media all’interno di ciascuna catena (\(W\)). La formula è la seguente:
\[ \widehat{R} = \sqrt{\frac{W + \frac{B}{n}}{W}}, \] dove \(n\) è il numero di campioni per catena. Un \(\widehat{R}\) vicino a 1 indica che la varianza tra le catene è comparabile con quella all’interno delle catene, suggerendo che tutte le catene stanno campionando dalla medesima distribuzione stazionaria.
N.6.1 Interpretazione e soglie
| Valore \(\widehat{R}\) | Interpretazione | Azione |
|---|---|---|
| 1.000 | Convergenza ottimale. | Nessuna. |
| ≤ 1.010 | Convergenza soddisfacente. | Monitorare se associato ad altri segnali. |
| 1.011 - 1.049 | Convergenza subottimale. | Verificare altri diagnostici e considerare azioni correttive. |
| ≥ 1.050 | Convergenza problematica. | Catene probabilmente non convergenti; intervento necessario. |
N.6.2 Implementazione in R
theta log_lik[1] log_lik[2] log_lik[3] log_lik[4] log_lik[5]
1.000875 1.000875 1.000875 1.000875 1.000875 1.000875
log_lik[6] log_lik[7] log_lik[8] log_lik[9] log_lik[10]
1.000875 1.000875 1.000875 1.000875 1.000875 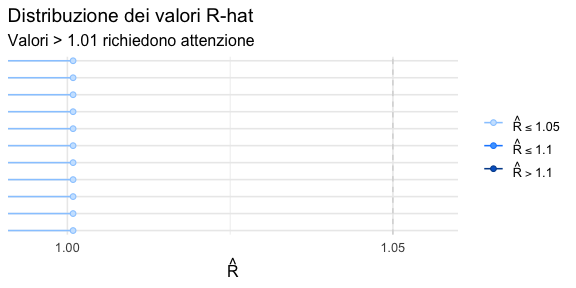
N.6.3 Azioni correttive per \(\widehat{R}\) elevato
Valori di \(\widehat{R}\) persistentemente superiori a 1.05 suggeriscono che le catene non hanno raggiunto una distribuzione stazionaria comune. Le strategie per affrontare il problema includono:
- Aumentare il numero di iterazioni, specialmente il periodo di warmup;
- Controllare e potenzialmente riparametrizzare il modello, poiché una cattiva geometria dello spazio dei parametri è una causa comune di non convergenza;
- Verificare le distribuzioni a priori e la coerenza dei dati per escludere problemi di identificazione o dati aberranti.
Un \(\widehat{R}\) accettabile è un prerequisito necessario, ma non sufficiente, per considerare valide le inferenze tratte dai campioni MCMC.
N.7 Diagnostica di Geweke
Il test di Geweke verifica l’ipotesi di stazionarietà di una catena MCMC confrontando la distribuzione dei campioni nella fase iniziale con quella nella fase finale. Nella sua implementazione standard, confronta la media dei primi 10% dei campioni con la media degli ultimi 50% dei campioni (escludendo il periodo di warmup). La differenza, standardizzata per l’errore standard, produce uno z-score.
N.7.1 Implementazione in R
# Preparazione dei dati: conversione in formato mcmc.list
df_theta <- as_draws_df(fit$draws("theta"))[, c(".chain", ".iteration", "theta")]
chains_list <- split(df_theta$theta, df_theta$.chain)
mcmc_list <- mcmc.list(lapply(chains_list, mcmc))
# Calcolo degli z-scores di Geweke per ciascuna catena
gd <- lapply(mcmc_list, geweke.diag, frac1 = 0.1, frac2 = 0.5)
z_scores <- sapply(gd, function(x) x$z)
cat("Z-scores di Geweke per catena:", round(z_scores, 3), "\n")Z-scores di Geweke per catena: -1.338 0.815 -0.583 0.915 # Calcolo dei p-value bilaterali associati
pvals <- 2 * pnorm(-abs(z_scores))
cat("P-value associati:", format.pval(pvals, digits = 3), "\n")P-value associati: 0.181 0.415 0.560 0.360 N.7.2 Interpretazione dei risultati
La logica del test è la seguente: se la catena è stazionaria, le medie delle due sottosezioni dovrebbero essere compatibili, producendo uno z-score vicino a zero.
- |z-score| < 1.96: non si rileva evidenza statistica di non-stazionarietà al livello di significatività del 5%.
- |z-score| ≥ 1.96: si osserva una differenza statisticamente significativa tra l’inizio e la fine della catena, suggerendo una possibile mancanza di stazionarietà (livello di significatività del 5%).
- |z-score| ≥ 2.58: evidenza forte di non-stazionarietà (livello dell’1%).
N.7.3 Considerazioni pratiche
Il test di Geweke è uno strumento diagnostico complementare. Un singolo test significativo può occasionalmente verificarsi per caso, specialmente quando si esaminano molti parametri. Tuttavia, z-score sistematicamente elevati (ad esempio, > 2) per diversi parametri chiave sono un segnale d’allarme che richiede un’indagine più approfondita, possibilmente ripetendo il campionamento con un maggior numero di iterazioni o intervenendo sulla parametrizzazione del modello.
N.8 Errore standard di Monte Carlo (MCSE)
L’errore standard di Monte Carlo (Monte Carlo Standard Error, MCSE) quantifica l’incertezza intrinseca nelle stime a posteriori derivante dal numero finito di campioni MCMC. A differenza della deviazione standard posteriore, che riflette l’incertezza statistica sul parametro, l’MCSE misura l’errore di simulazione introdotto dal processo di campionamento.
N.8.1 Definizione
Per una stima della media posteriore \(\bar{\theta}\), l’MCSE è definito come:
\[ \text{MCSE}(\bar{\theta}) = \frac{\hat{\sigma}_\theta}{\sqrt{N_{\text{eff}}}}, \] dove \(\hat{\sigma}_\theta\) è la deviazione standard posteriore stimata e \(N_{\text{eff}}\) è la dimensione effettiva del campione (Effective Sample Size). La formula evidenzia come l’MCSE diminuisca all’aumentare dell’informazione indipendente (\(N_{\text{eff}}\)) contenuta nella catena.
N.8.2 Calcolo e interpretazione in R
Errore standard di Monte Carlo per θ: 0.00175 L’MCSE dovrebbe essere almeno un ordine di grandezza inferiore alla deviazione standard posteriore corrispondente. In altre parole, l’incertezza dovuta al campionamento (MCSE) deve essere trascurabile rispetto all’incertezza statistica sulla stima del parametro. Un rapporto \(\text{SD}/\text{MCSE} \approx \sqrt{N_{\text{eff}}} > 10\) indica una precisione di simulazione adeguata.
N.8.3 Significato pratico
Un MCSE troppo elevato segnala che le stime dei parametri (come la media posteriore) potrebbero cambiare in modo sostanziale se si proseguisse il campionamento. Per ridurre l’MCSE è necessario aumentare \(N_{\text{eff}}\), il che si ottiene tipicamente prolungando la durata del campionamento, riducendo l’autocorrelazione attraverso una migliore parametrizzazione del modello, oppure aumentando il numero di catene parallele.
N.9 Transizioni divergenti nell’algoritmo HMC/NUTS
Le transizioni divergenti rappresentano un indicatore diagnostico critico nell’ambito del campionamento Hamiltonian Monte Carlo (HMC) e del suo algoritmo No-U-Turn Sampler (NUTS). Si verificano quando la traiettoria numerica, che approssima il sistema hamiltoniano, incontra regioni dello spazio dei parametri caratterizzate da una forte curvatura o da repentini cambiamenti di scala. In tali regioni, l’approssimazione discreta dell’equazione differenziale diventa instabile, portando a un significativo scostamento tra la traiettoria simulata e il percorso energetico vero, compromettendo la fedeltà del campionamento.
# Controllo del numero di transizioni divergenti
num_divergenze <- fit$diagnostic_summary()$num_divergent
cat("Numero di transizioni divergenti:", num_divergenze, "\n")Numero di transizioni divergenti: 0 0 0 0 N.9.1 Strategie di risoluzione
In presenza di divergenze, si raccomanda di intervenire secondo la seguente gerarchia:
-
Aumentare il parametro
adapt_delta: Questo parametro, compreso tra 0 e 1, controlla il livello di conservatorismo nell’adattamento della matrice di massa. Valori più elevati (ad esempio 0.95, 0.99) portano a una matrice di massa più conservativa e a step-size più piccoli, migliorando la stabilità a costo di una maggiore autocorrelazione.mod$sample(data = stan_data, adapt_delta = 0.99, ...) Riparametrizzare il modello: Molte problematiche geometriche sono dovute a parametrizzazioni “centrate” che creano forti dipendenze tra parametri. L’adozione di parametrizzazioni “non centrate” (non-centered parameterizations) può migliorare notevolmente la geometria dello spazio dei parametri, soprattutto per modelli gerarchici.
Rivedere le specifiche delle distribuzioni a priori: Prior eccessivamente informative o con supporto molto ristretto possono creare regioni di alta curvatura o “colli di bottiglia” che ostacolano l’esplorazione dello spazio dei parametri. Si consideri l’uso di prior più deboli o la trasformazione dei parametri.
Esaminare i dati osservati: Valori estremi o outlier nei dati possono causare regioni di alta curvatura nella distribuzione a posteriori. Un’analisi esplorativa preliminare e possibili trasformazioni dei dati possono mitigare il problema. Inoltre, verificare la coerenza delle scale delle variabili predittive.
Un numero elevato di transizioni divergenti segnala che il campionatore potrebbe non esplorare adeguatamente alcune regioni della distribuzione a posteriori, compromettendo la validità delle inferenze. La loro presenza richiede pertanto un’azione correttiva prima di procedere con l’analisi.
Un numero significativo di transizioni divergenti (> 0.1% delle iterazioni) indica che le stime potrebbero non essere affidabili.
N.10 Il BFMI (Bayesian Fraction of Missing Information)
Il BFMI è una metrica diagnostica che valuta l’efficienza dell’esplorazione dello spazio dei parametri da parte dell’algoritmo Hamiltonian Monte Carlo (HMC). Si basa sull’analisi delle fluttuazioni dell’energia del sistema hamiltoniano durante il campionamento. Valori bassi indicano che il campionatore fatica a esplorare alcune regioni dello spazio, tipicamente a causa di forti gradienti o di una geometria sfavorevole nella distribuzione a posteriori.
# Estrazione delle statistiche diagnostiche del sampler
sd_df <- fit$sampler_diagnostics(inc_warmup = FALSE, format = "df")
np_all <- sd_df |>
pivot_longer(
cols = c(accept_stat__, stepsize__, treedepth__, n_leapfrog__, divergent__, energy__),
names_to = "Parameter", values_to = "Value"
) |>
rename(Chain = .chain, Iteration = .iteration) |>
relocate(Chain, Iteration, Parameter, Value)
class(np_all) <- c("nuts_params", "data.frame")
np_energy <- np_all |>
dplyr::filter(Parameter == "energy__") |>
dplyr::select(Chain, Iteration, Parameter, Value)
class(np_energy) <- c("nuts_params", "data.frame")mcmc_nuts_energy(np_energy)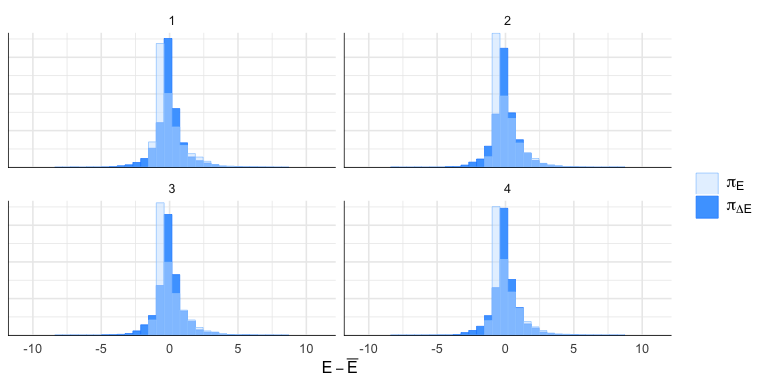
BFMI per catena: 1.097 1.209 1.112 1.088 cat("BFMI complessiva:", round(weighted.mean(bfmi, sapply(energy_by_chain, length)), 3), "\n")BFMI complessiva: 1.127 N.10.1 Interpretazione del BFMI
La seguente tabella fornisce una guida per l’interpretazione dei valori ottenuti:
| Valore BFMI | Interpretazione | Rischio |
|---|---|---|
| ≥ 0.3 | Esplorazione efficiente dello spazio dei parametri. | Basso |
| 0.2 – 0.3 | Esplorazione subottimale. Richiede attenzione e possibili interventi. | Moderato |
| < 0.2 | Esplorazione problematica. Le inferenze potrebbero essere inaffidabili. | Alto |
Un BFMI basso suggerisce spesso problemi nella parametrizzazione del modello (ad esempio, forti correlazioni tra parametri) o nella scelta delle distribuzioni a priori. Le strategie di intervento includono l’aumento del parametro adapt_delta, la riparametrizzazione del modello (ad esempio, utilizzando parametrizzazioni non centrate per effetti casuali), o la revisione della struttura del modello stesso.
N.11 LOO-CV e il parametro \(\kappa\) di Pareto
La Leave-One-Out Cross-Validation (LOO-CV) è un metodo per valutare la capacità predittiva out-of-sample di un modello bayesiano senza richiedere nuovi dati. L’implementazione efficiente utilizzata in Stan si basa sull’approssimazione Pareto-Smoothed Importance Sampling (PSIS), la cui affidabilità è monitorata tramite il parametro di forma \(\kappa\) della distribuzione di Pareto generalizzata.
N.11.1 Calcolo del LOO-CV in R
# Estrazione della log-verosimiglianza punto a punto
ll_array <- as_draws_array(fit$draws("log_lik"))
ll_mat <- as_draws_matrix(fit$draws("log_lik"))
# Parametri delle catene per il calcolo dell'efficienza relativa
S <- dim(ll_array)[1] # Numero di campioni per catena
C <- dim(ll_array)[2] # Numero di catene
chain_id <- rep(1:C, each = S)
# Calcolo dell'efficienza relativa
r_eff <- relative_eff(exp(ll_mat), chain_id = chain_id)
# Calcolo del LOO-CV con PSIS-LOO
loo_res <- loo::loo(ll_mat, r_eff = r_eff)
print(loo_res)
Computed from 16000 by 10 log-likelihood matrix.
Estimate SE
elpd_loo -7.1 1.3
p_loo 0.9 0.2
looic 14.2 2.6
------
MCSE of elpd_loo is 0.0.
MCSE and ESS estimates assume MCMC draws (r_eff in [0.4, 0.4]).
All Pareto k estimates are good (k < 0.7).
See help('pareto-k-diagnostic') for details.N.11.2 Diagnostica con il parametro \(\kappa\) di Pareto
Il parametro \(\kappa\) diagnostica la qualità dell’approssimazione PSIS per ciascuna osservazione. Valori elevati indicano che i pesi importance sampling hanno una distribuzione a code pesanti, rendendo la stima del LOO-CV instabile.
# Estrazione e riepilogo dei valori kappa
k_values <- pareto_k_values(loo_res)
cat("Riepilogo dei valori di Pareto kappa:\n")Riepilogo dei valori di Pareto kappa: Min. 1st Qu. Median Mean 3rd Qu. Max.
-0.001379 -0.001379 -0.001379 0.039054 0.099703 0.133397
Numero di osservazioni con kappa > 0.7: 0 N.11.3 Soglie interpretative
| Intervallo \(\kappa\) | Interpretazione | Raccomandazione |
|---|---|---|
| \(\kappa < 0.5\) | Approssimazione affidabile. | Le stime LOO-CV sono robuste. |
| \(0.5 \leq \kappa < 0.7\) | Approssimazione accettabile ma con qualche incertezza. | Interpretare con cautela. Controllare osservazioni specifiche. |
| \(\kappa \geq 0.7\) | Approssimazione potenzialmente inaffidabile. | Le stime potrebbero essere instabili. Considerare un modello più robusto o un cross-validation esplicito. |
N.11.4 Azioni correttive per \(\kappa\) elevato
La presenza di osservazioni con \(\kappa > 0.7\) (specialmente se > 1.0) suggerisce che il modello assegna una verosimiglianza estremamente bassa a quelle osservazioni. Si possono considerare:
- modelli più robusti (ad esempio, distribuzioni t-student invece di normali per i termini di errore);
- identificazione e analisi delle osservazioni influenti tramite un Pareto-k diagnostic plot;
- ricorso a una K-fold Cross-Validation esplicita per le osservazioni problematiche.
Il LOO-CV e il suo parametro \(\kappa\) forniscono quindi non solo una misura della performance predittiva, ma anche una diagnostica preziosa sull’adeguatezza del modello rispetto alla struttura dei dati osservati.
Prima di interpretare i risultati di un modello Stan, verificare sempre:
Riflessioni conclusive
La diagnostica MCMC non costituisce una fase opzionale o meramente tecnica, ma rappresenta una condizione necessaria per la validità delle inferenze bayesiane. Un modello, per quanto sofisticato nella sua formulazione teorica, produce risultati sostanzialmente inaffidabili se basato su catene che non hanno raggiunto la convergenza o che esplorano lo spazio dei parametri in modo inefficiente.
L’identificazione di problematiche attraverso gli strumenti diagnostici non è un fallimento, ma un’occasione per migliorare l’analisi. Le principali strategie correttive includono:
- estendere la durata del campionamento, aumentando le iterazioni totali e il periodo di warmup;
- ottimizzare la parametrizzazione del modello, adottando formulazioni che migliorino la geometria dello spazio dei parametri (ad esempio, parametrizzazioni non centrate per effetti casuali);
- rivedere la specificazione delle distribuzioni a priori, bilanciando l’informatività per evitare problemi di identificazione o regioni di curvatura eccessiva;
-
ottimizzare i parametri di controllo dell’algoritmo HMC/NUTS, come
adapt_deltaemax_treedepth; - verificare l’integrità e la coerenza dei dati osservati, poiché valori anomali o errori sistematici possono destabilizzare l’inferenza.
Nella comunicazione dei risultati, è buona pratica riportare in modo trasparente i valori chiave delle diagnostiche (quali \(\widehat{R}\), \(N_{\text{eff}}\), il conteggio delle divergenze e il BFMI). Tale trasparenza non solo rafforza la credibilità dell’analisi, ma favorisce anche la riproducibilità e consente una valutazione critica e costruttiva dei risultati ottenuti.
R version 4.5.2 (2025-10-31)
Platform: aarch64-apple-darwin20
Running under: macOS Tahoe 26.2
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
locale:
[1] C.UTF-8/UTF-8/C.UTF-8/C/C.UTF-8/C.UTF-8
time zone: Europe/Rome
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] tidyr_1.3.1 dplyr_1.1.4 loo_2.8.0 coda_0.19-4.1
[5] ggplot2_4.0.1 bayesplot_1.14.0 posterior_1.6.1 cmdstanr_0.8.0
loaded via a namespace (and not attached):
[1] tensorA_0.36.2.1 utf8_1.2.6 generics_0.1.4
[4] stringi_1.8.7 lattice_0.22-7 digest_0.6.39
[7] magrittr_2.0.4 evaluate_1.0.5 grid_4.5.2
[10] RColorBrewer_1.1-3 fastmap_1.2.0 plyr_1.8.9
[13] jsonlite_2.0.0 processx_3.8.6 backports_1.5.0
[16] ps_1.9.1 purrr_1.2.0 scales_1.4.0
[19] abind_1.4-8 cli_3.6.5 rlang_1.1.6
[22] withr_3.0.2 yaml_2.3.12 tools_4.5.2
[25] parallel_4.5.2 reshape2_1.4.5 checkmate_2.3.3
[28] vctrs_0.6.5 R6_2.6.1 matrixStats_1.5.0
[31] lifecycle_1.0.4 stringr_1.6.0 htmlwidgets_1.6.4
[34] pkgconfig_2.0.3 pillar_1.11.1 gtable_0.3.6
[37] glue_1.8.0 data.table_1.17.8 Rcpp_1.1.0
[40] xfun_0.54 tibble_3.3.0 tidyselect_1.2.1
[43] knitr_1.50 farver_2.1.2 htmltools_0.5.9
[46] labeling_0.4.3 rmarkdown_2.30 compiler_4.5.2
[49] S7_0.2.1 distributional_0.5.0